UCLA Scientists Develop New Algorithms to Study Genomic Data
Advance can further understanding of population structure to identify genetic risk factors for diseases
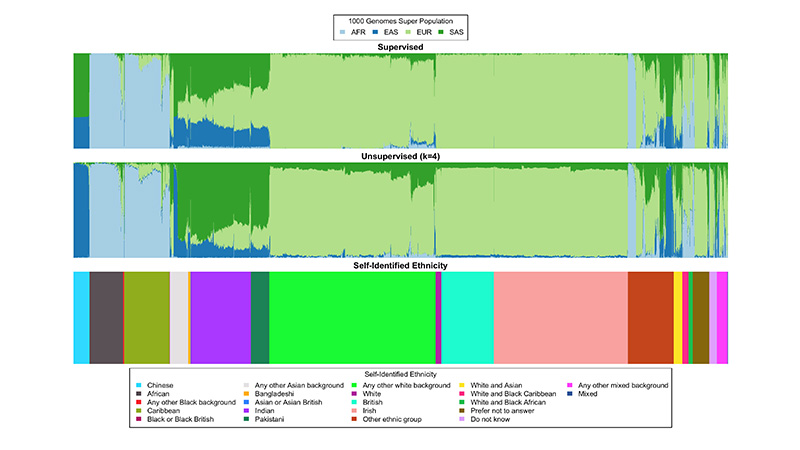
Courtesy of Sriram Sankararaman and Alec Chiu/UCLA
Individuals’ ancestry inferred by the UCLA-developed SCOPE computational method using UK Biobank data compared to their self-identified ethnicities
UCLA Samueli Newsroom
Led by UCLA associate professor Sriram Sankararaman, the research team has developed a computation method called SCalable pOPulation structure inferencE, or SCOPE, which is designed to speed up and scale inference of population structure. Population structure inference examines the genetic variations of a population to find patterns that guide conclusions or deter false associations in research.
The researchers made two major algorithmic improvements that enabled the team to analyze large genomic datasets accurately and expeditiously. Details on the new methods have recently been published in the American Journal of Human Genetics.
Sankararaman has joint appointments in UCLA’s departments of computer science and human genetics, as well as computational medicine — a department affiliated with both the UCLA Samueli School of Engineering and the David Geffen School of Medicine.
Since the early 2000s, there have been various methods used to study population structure, or comparisons of genomes in various populations, which can reveal how individuals relate to one another and to their ancestors. The findings are also key to identifying genetic risk factors for diseases that may affect a particular population more than others.
However, as the amount of genomic data has continued to grow rapidly over the years, improved computational methods that can keep pace with the massive growth of datasets are needed to provide fast and accurate genomic data analyses.
“We wanted to develop a method that would enable population structure inference, a commonly performed analysis, for the largest datasets available today and [the ones that are] yet to come,” said Alec Chiu.
“We wanted to develop a method that would enable population structure inference, a commonly performed analysis, for the largest datasets available today and [the ones that are] yet to come,” said Alec Chiu, the study’s lead author and a UCLA interdepartmental bioinformatics doctoral student studying under Sankararaman. “One of the important challenges in developing scalable methods is to provide improvements in terms of both time and computational resources.”
SCOPE improves upon previous population structure inference methods by reducing the amount of computational resources necessary to process large datasets, which also drives down the cost of running calculations. In one of its experiments, the research team used just 250 gigabytes of memory for computation instead of the roughly 2,000 gigabytes — the equivalent of 125 laptops with 16-GB memory each — needed with a previous research tool.
The research team has made SCOPE available to the public on its lab’s GitHub website, with the hope that it may benefit other researchers studying population genetics.
For their experiments, the researchers used UK Biobank’s repository of genomic data from 488,363 individuals, with many times more genetic variants.
“Several massive datasets are still yet to come, such as the All of Us Research Project and the Million Veteran Program, each of which aim to obtain genetic data from 1 million people,” Chiu said, referencing the datasets being compiled by the National Institutes of Health and the Department of Veterans Affairs, respectively. “We created SCOPE to enable analyses of such datasets.”
The interdisciplinary research team included individuals with experience in computer science, human genetics, machine learning and computational medicine from UCLA, Carnegie Mellon University, the University of Maryland and Meta Platforms, Inc.
Sankararaman’s lab also utilizes computational genetics to make discoveries about ancient human relatives, such as Denisovans, who were contemporaries of Neanderthals, and an unnamed extinct human relative that lived in West Africa.
The SCOPE research was supported by the National Institutes of Health, the National Science Foundation and an Alfred P. Sloan Research Fellowship.
Natalie Weber contributed to this story.