UCLA Researchers Develop Improved Computational Model to Analyze Antibody Patterns
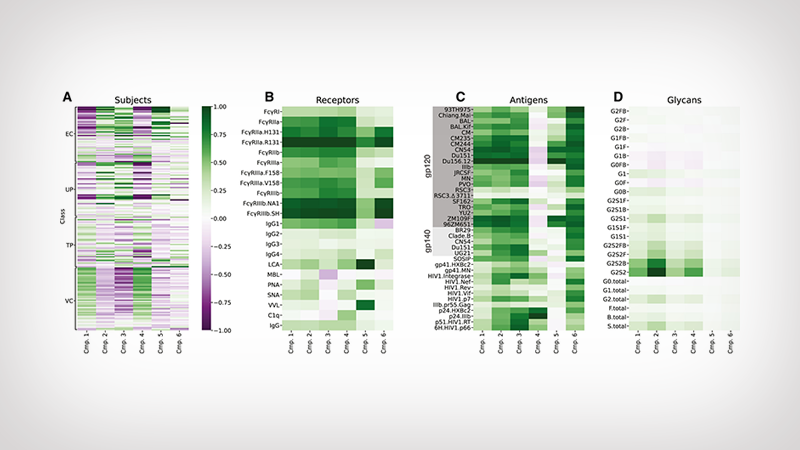
Visual depiction of the six patterns found in the HIV antibody analysis. Each plot shows how the patterns are represented among individuals (subjects); interactions with the immune system (receptors); parts of the HIV virus (antigens) and antibody molecular structure (glycans). The deeper the color, the stronger the correlation.
A systems serology study detailing the research findings was published in Molecular Systems Biology. Systems serology involves using experimental techniques to dissect antibodies’ features and functions, followed by a range of computational methods to mine through datasets to provide a better understanding of the interconnected relationships between the profiled antibodies and the response of the immune system.
The new computational method employed in this research simplifies the complex molecular interactions antibodies need to find and attach to viruses. It also accounts for how well the antibodies work and if they have adverse side effects. Thanks to the model’s ability to streamline collected data, vaccine specialists can find patterns in antibodies’ effectiveness more easily.
“This study shows how such antibody patterns can be greatly simplified and, in turn, help in the design of better therapies,” said Aaron Meyer.
“These datasets can be quite overwhelming as researchers who want to improve antibody-based treatments are faced with analyzing dozens of their molecular interactions, which can then result in secondary and tertiary reactions,” said research lead Aaron Meyer, an assistant professor of bioengineering at the UCLA Samueli School of Engineering. “This study shows how such antibody patterns can be greatly simplified and, in turn, help in the design of better therapies.”
Antibodies are Y-shaped proteins produced inside the body that identify and attach themselves to unwanted and often harmful objects, such as viruses and bacteria. Once connected to these pathogens using their two Y-shaped prongs, the antibodies’ Y base serves as a label recognizable to the immune system. The antibodies can either block the invading agents themselves or signal cells of the immune system, such as macrophages, to help eliminate the invaders.
The UCLA research helps determine whether an antibody’s therapeutic effects are due to how it binds to pathogens and destroys them, its role in activating cells in the immune system to attack the foreign object through its signaling base, or some combination of the two.
In their experiments, the researchers used a mathematical model called tensor decomposition to first build a matrix simulating disease progression before mining the data to identify otherwise difficult-to-find antibody patterns.
The researchers tested their methodology on antibody data collected from two separate groups.
First, the team examined data from 193 individuals who tested positive for COVID-19 that also showed how they fared, ranging from no symptoms to death. The UCLA model predicted whether the cases involving severe symptoms resulted in death with 74% accuracy.
The researchers found that the individuals who were hospitalized with severe symptoms but survived had a greater antibody response overall compared to those with moderate symptoms or others who died from the disease.
They also found that about 10% of the individuals had patterns of preexisting antibodies, which, according to the researchers, might have resulted from previous exposure to other types of coronaviruses.
For the second experiment, the team analyzed data collected from a group of 181 individuals infected with HIV. Some of them managed their infection with antiretroviral therapy while others managed their infection without therapy or got worse despite treatment. Using their model dissecting the antibody structure, the researchers correctly predicted nearly three-fourths of the patient outcomes against real data.
The lead author of the study is UCLA bioinformatics doctoral student Cyrillus Tan. Other authors, all from UCLA, are bioengineering undergraduate student Madeleine Murphy; Hakan Alpay, who recently graduated with a bachelor’s degree in computer science; and assistant bioengineering development engineer Scott Taylor.
The authors have made the computer code they developed available on GitHub.
Meyer is also a faculty member of the Jonsson Comprehensive Cancer Center at UCLA, and the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA.
The research was supported by a grant from the National Institutes of Health.